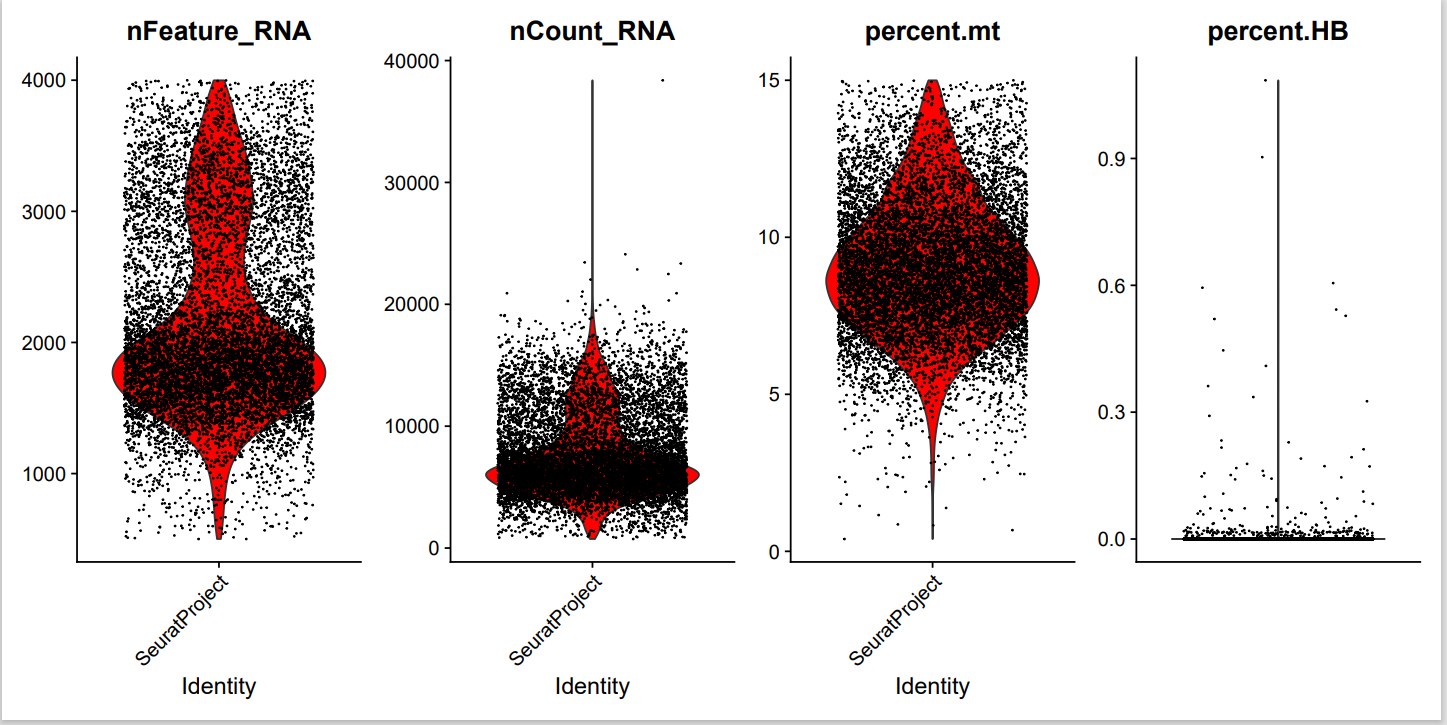
library(Seurat)
library(tidyverse)
library(patchwork)
rm(list=ls())
setwd('~/bioinformatics/scRNA')
dir.create("cluster")
scRNA <- readRDS("scRNA.rds")
scRNA <- FindVariableFeatures(scRNA, selection.method = "vst", nfeatures = 2000)
top10 <- head(VariableFeatures(scRNA), 10)
plot1 <- VariableFeaturePlot(scRNA)
plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE, size=2.5)
plot <- CombinePlots(plots = list(plot1, plot2),legend="bottom")
# ggsave("cluster/VariableFeatures.pdf", plot = plot, width = 8, height = 6)
# ggsave("cluster/VariableFeatures.png", plot = plot, width = 8, height = 6)
##如果内存足够最好对所有基因进行中心化
scale.genes <- rownames(scRNA)
scRNA <- ScaleData(scRNA, features = scale.genes)
##如果内存不够,可以只对高变基因进行标准化
# scale.genes <- VariableFeatures(scRNA)
# scRNA <- ScaleData(scRNA, features = scale.genes)
#原始表达矩阵
GetAssayData(scRNA,slot="counts",assay="RNA")
#标准化之后的表达矩阵
GetAssayData(scRNA,slot="data",assay="RNA")
#中心化之后的表达矩阵
GetAssayData(scRNA,slot="scale.data",assay="RNA")
# 细胞周期回归
CaseMatch(c(cc.genes$s.genes,cc.genes$g2m.genes),VariableFeatures(scRNA))
# 细胞周期评分
g2m_genes = cc.genes$g2m.genes
g2m_genes = CaseMatch(search = g2m_genes, match = rownames(scRNA))
s_genes = cc.genes$s.genes
s_genes = CaseMatch(search = s_genes, match = rownames(scRNA))
scRNA <- CellCycleScoring(object=scRNA, g2m.features=g2m_genes, s.features=s_genes)
# 查看细胞周期基因对细胞聚类的影响
scRNAa <- RunPCA(scRNA, features = c(s_genes, g2m_genes)) # scRNAa
p <- DimPlot(scRNAa, reduction = "pca", group.by = "Phase")
# ggsave("cluster/cellcycle_pca.png", p, width = 8, height = 6)
##如果需要消除细胞周期的影响
# scRNAb <- ScaleData(scRNA, vars.to.regress = c("S.Score", "G2M.Score"), features = rownames(scRNA))
scRNA <- RunPCA(scRNA, features = VariableFeatures(scRNA))
plot1 <- DimPlot(scRNA, reduction = "pca", group.by="orig.ident")
plot2 <- ElbowPlot(scRNA, ndims=20, reduction="pca")
plotc <- plot1+plot2
# ggsave("cluster/pca.pdf", plot = plotc, width = 8, height = 4)
# ggsave("cluster/pca.png", plot = plotc, width = 8, height = 4)
pc.num=1:18
# 获取PCA结果
# 此部分代码为分析非必须代码,不建议运行!!!
# 获取基因在pc轴上的投射值
Loadings(object = scRNA[["pca"]])
#获取各个细胞的pc值
Embeddings(object = scRNA[["pca"]])
#获取各pc轴解释量方差
Stdev(scRNA)
#查看决定pc值的top10基因, 此例查看pc1-pc5轴
print(scRNA[["pca"]], dims = 1:5, nfeatures = 10)
#查看决定pc值的top10基因在500个细胞中的热图,此例查看pc1-pc9轴
DimHeatmap(scRNA, dims = 1:9, nfeatures=10, cells = 500, balanced = TRUE)
# 细胞聚类
scRNA <- FindNeighbors(scRNA, dims = pc.num)
scRNA <- FindClusters(scRNA, resolution = 0.5)
table(scRNA@meta.data$seurat_clusters)
metadata <- scRNA@meta.data
cell_cluster <- data.frame(cell_ID=rownames(metadata), cluster_ID=metadata$seurat_clusters)
# write.csv(cell_cluster,'cluster/cell_cluster.csv',row.names = F)
# 非线性降维
#tSNE
scRNA = RunTSNE(scRNA, dims = pc.num)
embed_tsne <- Embeddings(scRNA, 'tsne')
# write.csv(embed_tsne,'cluster/embed_tsne.csv')
plot1 = DimPlot(scRNA, reduction = "tsne")
# ggsave("cluster/tSNE.pdf", plot = plot1, width = 8, height = 7)
# ggsave("cluster/tSNE.png", plot = plot1, width = 8, height = 7)
#UMAP
scRNA <- RunUMAP(scRNA, dims = 1:10)
embed_umap <- Embeddings(scRNA, 'umap')
# write.csv(embed_umap,'cluster/embed_umap.csv')
plot2 = DimPlot(scRNA, reduction = "umap")
# ggsave("cluster/UMAP.pdf", plot = plot2, width = 8, height = 7)
# ggsave("cluster/UMAP.png", plot = plot2, width = 8, height = 7)
#合并tSNE与UMAP
plotc <- plot1+plot2+ plot_layout(guides = 'collect')
# ggsave("cluster/tSNE_UMAP.pdf", plot = plotc, width = 10, height = 5)
# ggsave("cluster/tSNE_UMAP.png", plot = plotc, width = 10, height = 5)
##保存数据
# saveRDS(scRNA, file="scRNA_before_cluster.rds")